


CGPACK > JUN-2015 diary
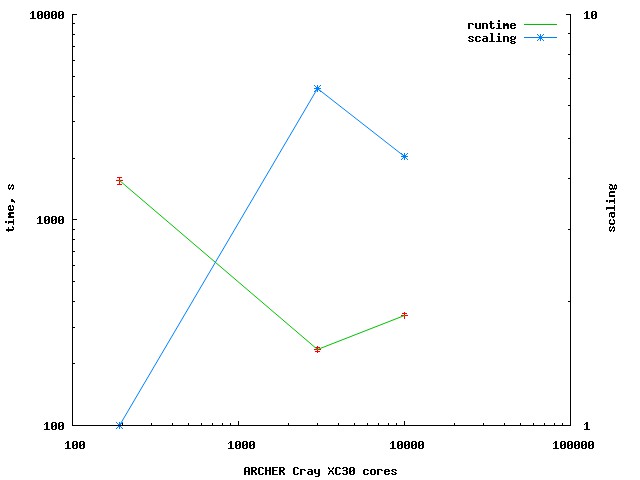
7-JUN-2015: Scaling and profiling results.
This data is from from
ARCHER.
The model uses 1M FE + 800M CA cells.
The program is
xx14noio.x.
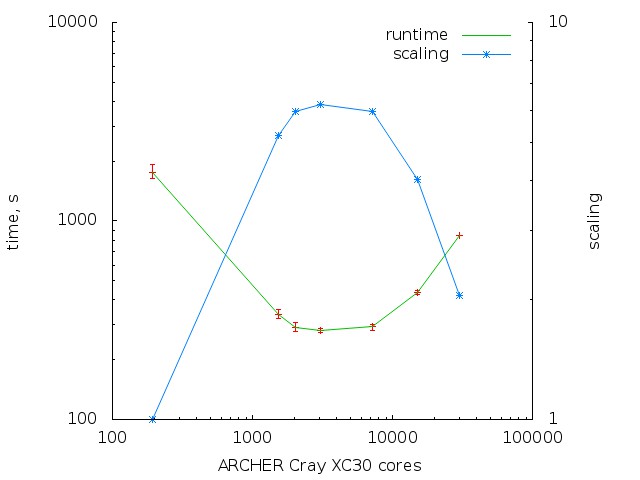
The scaling plot:
Profiling was done with CrayPAT S-2376-622 - Dec 2014, with default options:
pat_build -O apa xx14noio.x
This program has no CGPACK IO, hence the name.
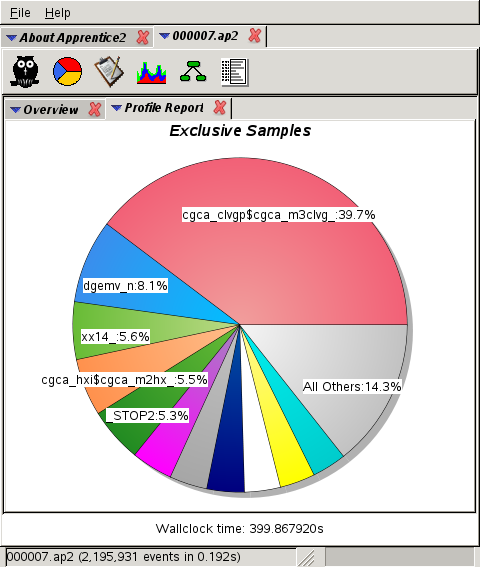
1,992 cores:
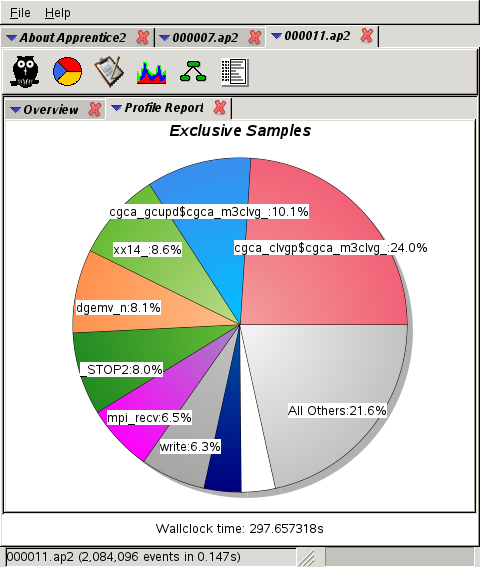
3,072 cores:
7,200 cores:
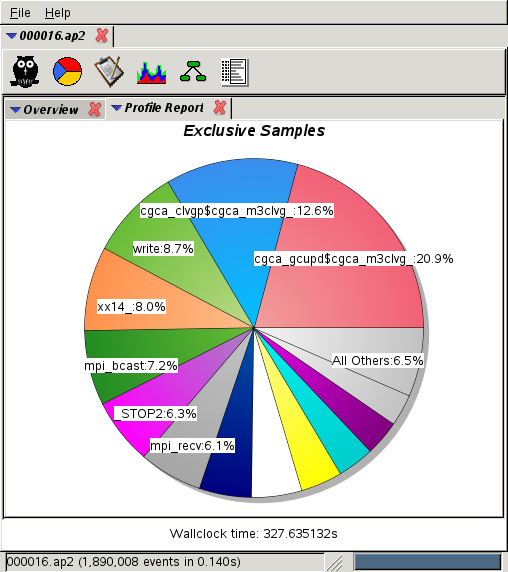
15,000 cores:
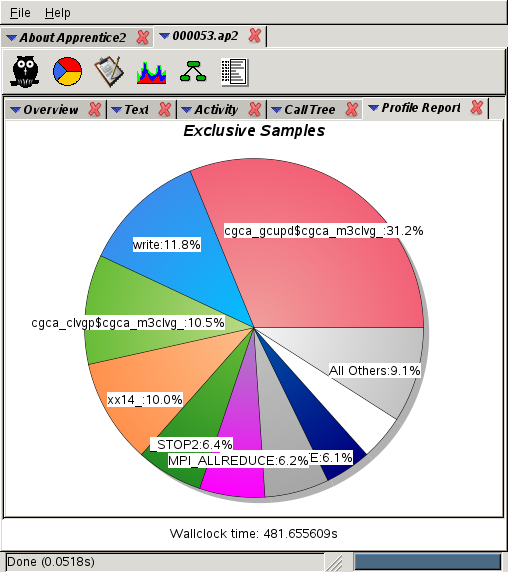
Conclusion:
cgca_gcupd,
routine that updates the grain
connectivity data is not scaling well.
This routine is not present at all at 2k cores,
takes 10% at 3k (second place),
21% at 7k cores (1st place) and
31% at 15k cores.
This routine is the prime candidate for optimisation.
8-JUN-2015: Porting CGPACK/ParaFEM to Intel 15 compiler.
A few links for the future:
User and Reference Guide for the Intel Fortran Compiler 15.0
Intel Parallel Studio XE 2015 Composer Edition Fortran Release Notes. Choose the linux version and the relevant update: initial release - ifort 15.0, update 1 - ifort 15.0.1, update 2 - 15.0.2, update 3 - 15.0.3, etc.
It seems there 2 ways of looking at MPI/coarray programming:
(1) an MPI program using coarrays or
(2) a coarray program using MPI calls.
Either way, with Intel,
MPI_FINALIZE
is *not* used.
My experience shows that path (1) does not work well.
Path (2) works like this:
program test
implicit none
include 'mpif.h'
integer :: rank, PEs, errstat, img, nimgs
! call MPI_INIT( errstat )
call MPI_COMM_SIZE( MPI_COMM_WORLD, PEs, errstat )
call MPI_COMM_RANK( MPI_COMM_WORLD, rank, errstat )
write (*,"(2(a6,i3))") "PEs:", PEs, "rank:", rank
img = this_image()
nimgs = num_images()
write (*,"(2(a6,i3))") "nimgs: ", nimgs, "img:", img
! call MPI_FINALIZE( errstat )
end program test
Note:
(1)
mpif.h
must be included.
(2) MPI environment is not explicitly set or destroyed!
This means
MPI_INIT
is *not* used.
This is done implicitly by Intel coarray runtime.
(3) The program is built as:
mpiifort -coarray=distributed -coarray-config-file=test.conf test.f90
where
mpiifort --version ifort (IFORT) 15.0.2 20150121 Copyright (C) 1985-2015 Intel Corporation. All rights reserved.
Then the program can be submitted to PBS queue
as a coarray program, i.e.
mpirun
is not needed!
Again, remember that all this is relevant only
for Intel environment:
#!/bin/sh #$Id: pbs.bc3 1868 2015-06-04 22:40:52Z shterenlikht@gmail.com $ #PBS -l walltime=00:02:00,nodes=2:ppn=16 #PBS -j oe cd $HOME/parafem/parafem/src/programs/dev/xx14 export OUTFILE=test.out echo > $OUTFILE echo "LD_LIBRARY_PATH: " $LD_LIBRARY_PATH >> $OUTFILE echo "which mpirun: " `which mpirun` >> $OUTFILE export I_MPI_DAPL_PROVIDER=ofa-v2-ib0 >> $OUTFILE mpdboot --file=$PBS_NODEFILE -n 1 >> $OUTFILE mpdtrace -l >> $OUTFILE echo "START TIME:" `date` >> $OUTFILE #mpirun -n 64 xx14std.x p121_medium >> $OUTFILE a.out >> $OUTFILE mpdallexit >> $OUTFILE echo "END TIME: " `date` >> $OUTFILE
Finally, file
test.conf
contains:
-envall -n 20 a.out
The (sorted) output looks like this:
PEs: 20 rank: 0 Thus no job control in this shell. Warning: no access to tty (Bad file descriptor). nimgs: 20 img: 1 PEs: 20 rank: 1 nimgs: 20 img: 2 PEs: 20 rank: 2 nimgs: 20 img: 3 PEs: 20 rank: 3 nimgs: 20 img: 4 PEs: 20 rank: 4 nimgs: 20 img: 5 PEs: 20 rank: 5 nimgs: 20 img: 6 PEs: 20 rank: 6 nimgs: 20 img: 7 PEs: 20 rank: 7 nimgs: 20 img: 8 PEs: 20 rank: 8 nimgs: 20 img: 9 PEs: 20 rank: 9 nimgs: 20 img: 10 PEs: 20 rank: 10 nimgs: 20 img: 11 PEs: 20 rank: 11 nimgs: 20 img: 12 PEs: 20 rank: 12 nimgs: 20 img: 13 PEs: 20 rank: 13 nimgs: 20 img: 14 PEs: 20 rank: 14 nimgs: 20 img: 15 PEs: 20 rank: 15 nimgs: 20 img: 16 PEs: 20 rank: 16 nimgs: 20 img: 17 PEs: 20 rank: 17 nimgs: 20 img: 18 PEs: 20 rank: 18 nimgs: 20 img: 19 PEs: 20 rank: 19 nimgs: 20 img: 20
Note that I asked for 32 processors (PEs), but used only 20.
Note that this statement:
lcentr = (/ lcentr, mcen( img_curr, j, cen_ca ) /)
from
cgca_m2pfem/cgca_pfem_cenc
requires ifort flag
-assume realloc_lhs
to be understood correctly. It took me a day of debugging to figure this out. See Intel Fortran 15 ref guide.
9-JUN-2015: More on porting ParaFEM/CGPACK to Intel 15
Seems I got it.
This the deformed FE mesh from a coupled ParaFEM/CGPACK run.
The model is p121_medium.
The CGPACK was run with a reduced resolution of 10,000 cells per grain,
to speed up the run.
There are 1000 grains in the model.
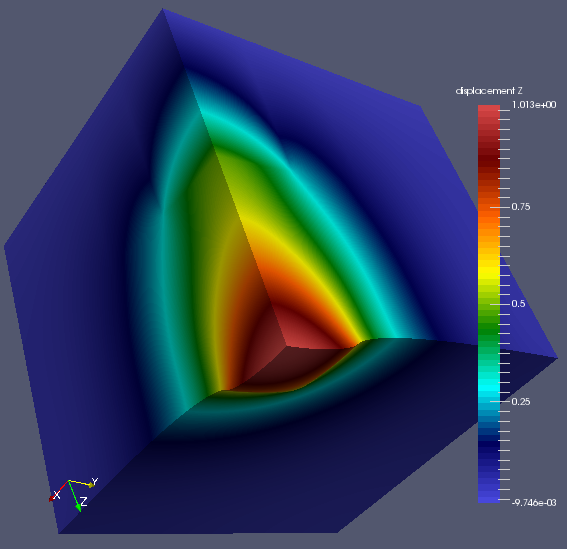
On the CA level the model looks like this. (Something is wrong with paraview colours):
Cracks:

Grain boundries:

Cracks with semi-transparent GB:

11-JUN-2015: Still playing with Intel 15
For the future - Cray Fortran Reference Manual: S-3901-83 - May 2014.
24-JUN-2015: Slightly improved scaling
Following profiling,
cgca_gcupd
was optimised to make remote comms more
evenly spread.
Also, several
sync all
have been removed from
xx14noio
program.
As a result, the scaling from 200 to 3k cores
improved slightly.
Scaling to 10k cores is still bad.
Need to profile again.
6-APR-2016: Raw timing data
Just found the raw timing data used for the above plots:
timing_large